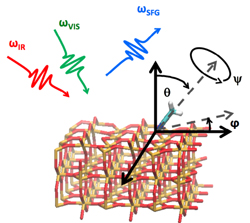
In recent years, we have especially been interested in modelling of spectra for condensed phase systems. Besides studies for the inclusion of solvent effects in static calculations, we have focused on ab initio molecular dynamics as a tool to describe condensed phase systems and their dynamics at ambient conditions. Apart from the use of density functional theory embedding, we have, for instance, presented efficient computational approaches for Raman (optical activity) and sum frequency generation. Functional solvated molecules and ionic liquids have been studied together with the group of T. Bürgi (University of Geneva).
Selected Publications
- Luber, S. Solvent Effects in Calculated Vibrational Raman Optical Activity Spectra of α-Helices J. Phys. Chem. A 2013, 117, 2760-2770.
- Luber, S.; Iannuzzi, M.; Hutter, J. Raman spectra from ab initio molecular dynamics and its application to liquid S-methyloxirane J. Chem. Phys. 2014, 141, 094503.
- Luber, S. Local Electric Dipole Moments for Molecular Periodic Systems via Density Functional Theory Embedding J. Chem. Phys. 2014, 141, 234110.
- Luber, S. Sum frequency generation of acetonitrile on rutile (110) surface from density functional theory-based molecular dynamics J. Phys. Chem. Lett. 2016, 7, 5183-5187.
- Oulevey, P.; Luber, S.; Varnholt, B.; Bürgi, T. Symmetry Breaking in Chiral Ionic Liquids Evidenced by Vibrational Optical Activity Angew. Chem. Int. Ed. 2016, 55, 11787-11790.
- Mališ, M., Luber, S. Trajectory Surface Hopping Nonadiabatic Molecular Dynamics with Kohn–Sham ΔSCF for Condensed-Phase Systems J. Chem. Theory Comput., 2020, 16, 7, 4071-4086